



EnviroPouch® Pouch Testing Compliance
 Enviropak® LLC dba EnviroPouch® maintains annual US FDA establishment registration, #1056276, medical device class II. The US FDA 510(k) Premarket Notification clearance to market EnviroPouch® product is registered as ProPak, #K924118. Think of the 510(k) as the EnviroPouch® “birth certificate;” The 510(k) related to EnviroPouch® information does not change over time, although the product, company, and/or ownership names may change.
Enviropak® LLC dba EnviroPouch® maintains annual US FDA establishment registration, #1056276, medical device class II. The US FDA 510(k) Premarket Notification clearance to market EnviroPouch® product is registered as ProPak, #K924118. Think of the 510(k) as the EnviroPouch® “birth certificate;” The 510(k) related to EnviroPouch® information does not change over time, although the product, company, and/or ownership names may change.
CONSTRUCTION
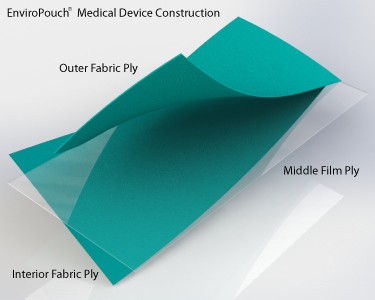 EnviroPouch® is constructed of a multi-layered fabric and film-barrier design. The film barrier design of the EnviroPouch® uniquely defines the first and only reusable steam sterilization pouch in the world since 1993 to clinically demonstrate the ability to achieve and maintain sterility to meet CDC guidelines for infection control and to adhere to the US FDA requirements for a medical device class II.
EnviroPouch® is constructed of a multi-layered fabric and film-barrier design. The film barrier design of the EnviroPouch® uniquely defines the first and only reusable steam sterilization pouch in the world since 1993 to clinically demonstrate the ability to achieve and maintain sterility to meet CDC guidelines for infection control and to adhere to the US FDA requirements for a medical device class II.
CDC Guidelines
CDC Guidelines for Infection Control in Dental Health-Care Settings
EnviroPouch medical device construction is consistent with and meets or exceeds CDC guidelines.
See "Packaging and Preparation" section.
EnviroPouch® Instructions for Use
How is the accreditation process impacted by our utilization of Enviropouch®, if at all?
For 30 years, Enviropouch® continually achieves the following:
1) US FDA medical device class II facility registration, with premarket approval,
2) comprehensive successful FDA audits of our facility,
3) meeting or exceeding CDC guidelines for infection control in healthcare and other professional steam infection control settings,
4) formal accreditations of our customers with their utilization of Enviropouch®,
5) international utilization of Enviropouch® with original focus in US dental and dental hygiene colleges with their infection control departments' utilization of Enviropouch®.
NOTE: If you encounter misinformation about the US FDA to do with "approval", please be educated here about this common, long term, false advertising practice to 1) attempt to fool the public into purchasing products marketed in this manner, or 2) specifically in our case, use this misinformation or characterization about a medical device product as a competitive misinformation ploy, often characterized as libel, intended by an industry competitor to disparage and discourage purchasing of a competitor's products with false, misleading misinformation that would suggest a medical device itself is approved or not approved by the FDA. The truth is a US medical device manufacturing facility may be registered or not, and the facility may or may not have premarket approval to be marketed or not.
Please utilize this US FDA link to educate yourself and professional peers. https://www.fda.gov/consumers/consumer-updates/it-really-fda-approved



.gif "EnviroPouch® accepts Visa, Mastercard, Discover and American Express credit cards")